
53 Best Bodyweight Crossfit Workouts to Stay Fit with No Equipment
Whether you’re at home, traveling, or in a beautiful park somewhere, bodyweight workouts are a…

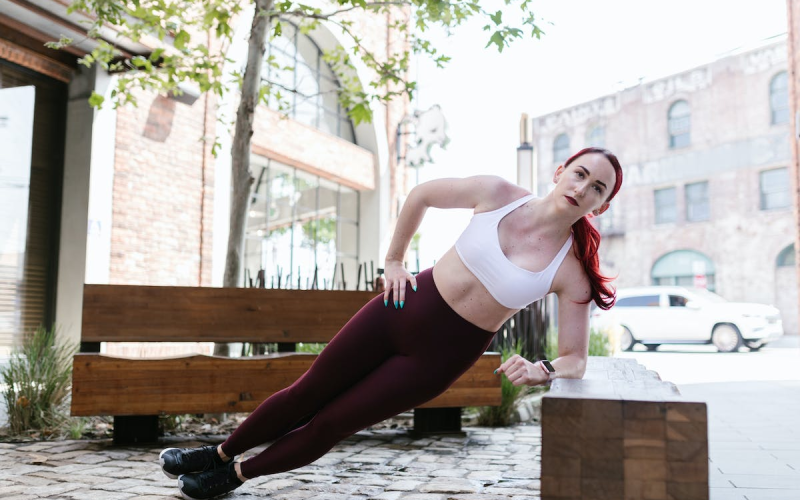
The Best Crossfit Bodyweight Exercises
Crossfit has become a popular fitness routine that is quickly growing in popularity. It’s no…
